#animal model

The active ingredient in an over-the-counter skin cream might do more than prevent wrinkles. Scientists have discovered that the drug, called kinetin, also slows or stops the effects of Parkinson’s disease on brain cells.
Scientists identified the link through biochemical and cellular studies, but the research team is now testing the drug in animal models of Parkinson’s. The research is published in the August 15, 2013 issue of the journal Cell.
“Kinetin is a great molecule to pursue because it’s already sold in drugstores as a topical anti-wrinkle cream,” says HHMI investigator Kevan Shokat of the University of California, San Francisco. “So it’s a drug we know has been in people and is safe.”
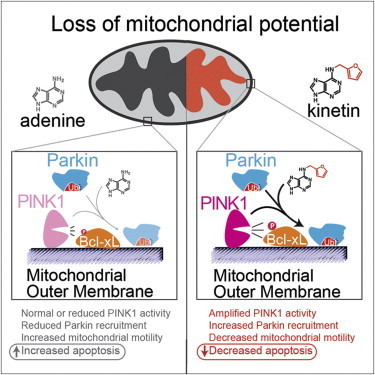
Parkinson’s disease is a degenerative disease that causes the death of neurons in the brain. Initially, the disease affects one’s movement and causes tremors, difficulty walking, and slurred speech. Later stages of the disease can cause dementia and broader health problems. In 2004, researchers studying an Italian family with a high prevalence of early-onset Parkinson’s disease discovered mutations in a protein called PINK1 associated with the inherited form of the disease.
Since then, studies have shown that PINK1 normally wedges into the membrane of damaged mitochondria inside cells that causes another protein, Parkin, to be recruited to the mitochondria, which are organelles responsible for energy generation. Neurons require high levels of energy production, therefore when mitochondrial damage occurs, it can lead to neuronal death. However, when Parkin is present on damaged mitochondria, studding the mitochondrial surface, the cell is able to survive the damage. In people who inherit mutations in PINK1, however, Parkin is never recruited to the organelles, leading to more frequent neuronal death than usual.
Shokat and his colleagues wanted to develop a way to turn on or crank up PINK1 activity, therefore preventing an excess of cell death, in those with inherited Parkinson’s disease. But turning on activity of a mutant enzyme is typically more difficult than blocking activity of an overactive version.
“When we started this project, we really thought that there would be no conceivable way to make something that directly turns on the enzyme,” says Shokat. “For any enzyme we know that causes a disease, we have ways to make inhibitors but no real ways to turn up activity.”
His team expected it would have to find a less direct way to mimic the activity of PINK1 and recruit Parkin. In the hopes of more fully understanding how PINK1 works, they began investigating how PINK1 binds to ATP, the energy molecule that normally turns it on. In one test, instead of adding ATP to the enzymes, they added different ATP analogues, versions of ATP with altered chemical groups that slightly change its shape. Scientists typically must engineer new versions of proteins to be able to accept these analogs, since they don’t fit into the typical ATP binding site. But to Shokat’s surprise, one of the analogs—kinetin triphosphate, or KTP—turned on the activity of not only normal PINK1, but also the mutated version, which doesn’t bind ATP.
“This drug does something that chemically we just never thought was possible,” says Shokat. “But it goes to show that if you find the right key for the right lock, you’ll be able to open the door.”
To test whether the binding of KTP to PINK1 led to the same consequences as the usual ATP binding, Shokat’s group measured the activity of PINK1 directly, as well as the downstream consequences of this activity, including the amount of Parkin recruited to the mitochondrial surface, and the levels of cell death. Adding the precursor of KTP, kinetin, to cells—both those with PINK1 mutations and those with normal physiology—amplified the activity of PINK1, increased the level of Parkin on damaged mitochondria, and decreased levels of neuron death, they found.
“What we have here is a case where the molecular target has been shown to be important to Parkinson’s in human genetic studies,” says Shokat. “And now we have a drug that specifically acts on this target and reverses the cellular causes of the disease.”
The similar results in cells with and without PINK1 mutations suggest that kinetin, which is a precursor to KTP, could be used to treat not only Parkinson’s patients with a known PINK1 mutation, but to slow progression of the disease in those without a family history by decreasing cell death.
Shokat is now performing experiments on the effects of kinetin in mice with various forms of Parkinson’s disease. However, the usefulness of animal models in Parkinson’s research has been debated, and therefore the positive results from the cellular data, he says, is as good an indicator as results in animals that this drug has potential to treat Parkinson’s in humans. Initial human studies will likely focus on the small population of patients with PINK1 mutations, and if successful in that group the drug could later be tested in a wider array of Parkinson’s patients.

Mouse study finds link between gut bacteria and neurogenesis
Antibiotics strong enough to kill off gut bacteria can also stop the growth of new brain cells in the hippocampus, a section of the brain associated with memory, reports a study in mice published May 19 in Cell Reports. Researchers also uncovered a clue to why– a type of white blood cell seems to act as a communicator between the brain, the immune system, and the gut.
“We found prolonged antibiotic treatment might impact brain function,” says senior author Susanne Asu Wolf of the Max-Delbrueck-Center for Molecular Medicine in Berlin, Germany. “But probiotics and exercise can balance brain plasticity and should be considered as a real treatment option.”
Wolf first saw clues that the immune system could influence the health and growth of brain cells through research into T cells nearly 10 years ago. But there were few studies that found a link from the brain to the immune system and back to the gut.
In the new study, the researchers gave a group of mice enough antibiotics for them to become nearly free of intestinal microbes. Compared to untreated mice, the mice who lost their healthy gut bacteria performed worse in memory tests and showed a loss of neurogenesis (new brain cells) in a section of their hippocampus that typically produces new brain cells throughout an individual’s lifetime. At the same time that the mice experienced memory and neurogenesis loss, the research team detected a lower level of white blood cells (specifically monocytes) marked with Ly6Chi in the brain, blood, and bone marrow. So researchers tested whether it was indeed the Ly6Chi monocytes behind the changes in neurogenesis and memory.
In another experiment, the research team compared untreated mice to mice that had healthy gut bacteria levels but low levels of Ly6Chi either due to genetics or due to treatment with antibodies that target Ly6Chi cells. In both cases, mice with low Ly6Chi levels showed the same memory and neurogenesis deficits as mice in the other experiment who had lost gut bacteria. Furthermore, if the researchers replaced the Ly6Chi levels in mice treated with antibiotics, then memory and neurogenesis improved.
“For us it was impressive to find these Ly6Chi cells that travel from the periphery to the brain, and if there’s something wrong in the microbiome, Ly6Chi acts as a communicating cell,” says Wolf.
Luckily, the adverse side effects of the antibiotics could be reversed. Mice who received probiotics or who exercised on a wheel after receiving antibiotics regained memory and neurogenesis. “The magnitude of the action of probiotics on Ly6Chi cells, neurogenesis, and cognition impressed me,” she says.
But one result in the experiment raised more questions about the gut’s bacteria and the link between Ly6Chi and the brain. While probiotics helped the mice regain memory, fecal transplants to restore a healthy gut bacteria did not have an effect.
“It was surprising that the normal fecal transplant recovered the broad gut bacteria, but did not recover neurogenesis,” says Wolf. “This might be a hint towards direct effects of antibiotics on neurogenesis without using the detour through the gut. To decipher this we might treat germ free mice without gut flora with antibiotics and see what is different.”
In the future, researchers also hope to see more clinical trials investigating whether probiotic treatments will improve symptoms in patients with neurodegenerative and psychiatric disorders.“We could measure the outcome in mood, psychiatric symptoms, microbiome composition and immune cell function before and after probiotic treatment,” says Wolf.
Post link

(Image caption: Lines labeling cortical subplate, mesencephalic, and diencephalic cell types (see Fig. 7 in Shima et al.))
Trapping individual cell types in the mouse brain
The complexity of the human brain depends upon the many thousands of individual types of nerve cells it contains. Even the much simpler mouse brain probably contains 10,000 or more different neuronal cell types. Brandeis scientists Yasu Shima, Sacha Nelson and colleagues report in the journal eLife on a new approach for genetically identifying and manipulating these cell types.
Cells in the brain have different functions and therefore express different genes. Important instructions for which genes to express, in which cell types, lie not only in the genes themselves, but in small pieces of DNA called enhancers found in the large spaces between genes. The Brandeis group has found a way to highjack these instructions to express other artificial genes in particular cell types in the mouse brain. Some of these artificially expressed genes (also called transgenes) simply make the cells fluorescent so they can be seen under the microscope. Other transgenes are master regulators that can be used to turn on or off any other gene of interest. This will allow scientists to activate or deactivate the cells to see how they alter behavior, or to study the function of specific genes by altering them only in some cell types without altering them everywhere in the body. In addition to developing the approach, the Brandeis group created a resource of over 150 strains of mice in which different brain cell types can be studied.
Post link
A team of scientists has identified the existence of a back-up plan for memory storage, which comes into play when the molecular mechanism of primary long-term memory storage fails.

Previous work had shown that mice engineered without an enzyme crucial to long-term memory storage could still form such memories, creating a controversy that a team of scientists has now resolved with the new research, which appears in the journal eLife.
The research focused on an enzyme made in nerve cells—PKMzeta. In a series of experiments, they confirmed that while the enzyme is crucial to long-term memory in normal mice, the mice engineered without PKMzeta still form long-term memories because they deploy an alternative, previously silent memory-storage method.
“Mice missing the PKMzeta enzyme essential for long-term memory are able to recruit a back-up mechanism for long-term memory storage,” explains André Fenton, a professor in NYU’s Center for Neural Science and one of the paper’s co-authors. “The question now is: how does PKMzeta function and what is the mechanism of its interaction with the PKCiota/lambda backup mechanisms?”
Previous research has found PKMzeta plays an important role in long-term memory storage, which scientists believe depends on the persistent strengthening of the connections between nerve cells. Specifically, the enzyme is made during the strengthening of these connections, and it remains in place so long as the links remain strong.
Notably, studies examining the role of PKMzeta’s in memory found that when the enzyme’s function was weakened in rodents,after they formed long-term memory, the animals could no longer remember diverse types of memories depending on diverse parts of the brain, suggesting that PKMzeta is a general memory storage mechanism.
But, recently, the importance of PKMzeta was questioned by experiments on genetically engineered “knockout” mice in which the gene that makes PKMzeta was deleted. Without PKMzeta, the knockout mice could still strengthen connections between nerve cells and still learn and remember.
While some took these results as evidence that PKMzeta’s role had previously been overstated, such studies did not consider the possibility of a “back-up” mechanism for memory that takes over when PKMzeta is removed. So the question remained: Is PKMzeta unimportant for memory or, in its absence, is a back-up mechanism deployed?
To address this matter, Panayiotis Tsokas, a research professor in Todd Sacktor’s lab at SUNY Downstate Medical Center, Fenton, and their colleagues tested both the “PKMzeta is unimportant” and “PKMzeta is compensated” hypotheses. To do so, they used a piece of modified DNA as a drug to block the formation of PKMzeta. If another molecule or molecules act as a back-up mechanism for PKMzeta, the scientists reasoned, the new drug would block the formation of memory in normal mice, but would have no effect on memory in the knockout mice that cannot make PKMzeta — the drug would have nothing to work on.
The results supported the PKMzeta is compensated hypothesis—the formation of memories normal mice was disrupted while that for the knockout mice was not, confirming the importance of PKMzeta, but also pointing to the presence of a back-up mechanism, which they identified involves PKCiota/lambda, the most closely related molecule to PKMzeta.